Archaeological context of Kadruka 1 SK68
Archaeological fieldwork in the Kadruka district of northern Sudan has revealed widespread past occupation of the alluvial plain and paleochannel systems to the east of the current Nile River channel9. In accordance with broader archaeological sequences for this region of the Middle Nile Valley10,11,12, cultural deposits were primarily linked to the Neolithic (spanning the 7th millennium BP), as well as the more recent Kerma period (3450–4450 BP). Excavation programs in the Kadruka district focused on several cemeteries from these cultural periods, including Kadruka 1 and Kadruka 21. Reflecting local environmental fluctuations between dry and wet conditions associated with more intensive Nile floods during the Middle Holocene, Neolithic skeletal remains at these cemeteries are typically highly degraded13,14. In contrast, progressive local aridification and floodplain contraction15, has facilitated enhanced preservation of organics, including hair and leather items, in the more recent Kerma period burials at Kadruka 116.
A recent study exploring the diet of individuals from Kadruka 21 and Kadruka 1 detected milk proteins attributable to domesticated cow (Bovinae) or sheep (Ovis) in the dental calculus of individual SK129 from Kadruka 21 and to goat (Carpa) in the dental calculus of individual SK68 from Kadruka 117. Furthermore, isotopic analyses of hair from individual SK68 (named “Sudan_Kadruka1_4000BP” in this study) broadly indicate a diet primarily composed of C3-based resources (C3 plants or animals consuming C3 plants, δ13C -17.0‰, δ15N 12.0‰).
The early proliferation of herding economies in northeastern Africa, particularly apparent in the Kerma civilisation of Upper Nubia11,12, has been proposed as a potential link to the dispersal of pastoral populations into eastern Africa5,6,7, though there is as yet no published genetic evidence to support this migration model. Direct palaeoproteomic evidence for milk consumption17, together with the remains of cattle, sheep and goat found in grave assemblages18, identify individuals from Kadruka 1 and Kadruka 21 as belonging to early populations practicing pastoralism, making the sites ideal for archaeogenetic research seeking to examine early pastoralist dispersals in northeastern Africa.
Ancient DNA authentification and sequencing strategy
We screened five specimens deriving from four individuals from the Kadruka district of northern Sudan (Fig. 1a) for aDNA preservation, drawing on tooth (KDR001.A), hair (KDR001.B), petrous bone (KDR002, KDR004) and cranial (KDR003) samples. All specimens were excavated from archaeological contexts dating to the Neolithic and Kerma periods. The only sample that yielded detectable authentic aDNA was a lock of dark hair (127 mg) (Fig. 1b) from a Kerma period individual. We used a total of 27.5 cm of hair for aDNA extraction and 78.5 mg for radiocarbon dating. The hair sample from Kadruka 1 SK68 is directly dated to 3928–4139 calBP (Fig. 1c), contemporaneous to the Pastoral Neolithic period in eastern Africa.
Sample details of the 4,000-year-old hair from Sudan. (a) Geographic location of Kadruka and modern African populations used for subsequent analyses. (b) Picture of the Kadruka hair sample. (c) C14 calibrated age (cal BC) of the Kadruka hair, plotted using IntCal13 calibration curve19.
To maximize the possibility of aDNA retrieval from shotgun sequencing, we applied double-stranded and single-stranded library protocols to the four extracts deriving from the Kadruka skeletal remains, but only applied the single-stranded library protocol to the hair sample, since this protocol typically results in higher yields than the double-stranded library approach for highly fragmented DNA. None of the aDNA libraries extracted from the skeletal elements showed authentic aDNA damage patterns, i.e., substitutions from cytosine to thymine (C- > T) (Table S1). The hair sample (KDR001.B0101), however, provided an observed 17.5% C- > T substitution rate at the first 5-prime base in sequencing reads (Table S1). Accordingly, we selected this library from the hair sample for deeper sequencing.
We found that the average read length was relatively short (33 base pair/bp, Table S1). This poses a challenge, since short DNA fragments can result in spurious alignments to the human reference genome, even if they do not originate from humans, but from, for example, microbes present in the burial environment. On the other hand, while long DNA fragments have high mapping certainty, they are more likely to originate from modern human DNA contamination (which typically consists of long DNA fragments). We therefore explored various read length cut-offs to yield as much authentic human aDNA as possible, while maintaining a low proportion of reads from presumed modern human contamination. For this purpose, we used two tools to assess both the rate of spurious alignments and the rate of modern human contaminants. First, we used SpAl20 which uses simulations to estimate fractions of spurious and authentic alignments given certain read length cut-offs. For a cut-off length of 25 bp, SpAI estimated a spurious alignment fraction of 10% (Table S2). As the read length cut-off increases, the estimation for spurious alignments drops respectively (Table S2). Second, we used AuthentiCT21 to estimate the overall contamination level in the aligned fragments using base substitution patterns. We explored length cut-offs at 10, 25, 30 (custom setting) and 34 bp in the raw data processing steps, and summarised EAGER statistics and respective contamination estimates in Table S2.
We find that at a length cut-off of 30 bp, 47.3 ± 2.4% of retrieved aDNA is likely of modern-human contaminant origin (Table 1). In comparison, a length cut-off of 25 bp yielded 4,680,356 mapped reads with 0.1 ± 0.3% contamination (Table 1). Together with the results from SpAL, we consider 25 bp a safe cut-off length for this library. Thus, we continued our downstream analyses with a 25 bp read length filter, ending up with 231,040 sequencing reads after mapping, from which we derived 3,336 pseudo-haploid allele calls on 1240 k SNP positions (Table 1).
Characteristics of the aDNA fragments from hair
Employing our final read length filter at 25 bp and additionally filtering for alignment mapping quality (Methods), we further explored alignment statistics. We find two unusual characteristics in the aDNA library generated from the Kadruka hair sample. The first is that the sample is enriched in unusually short DNA molecules, giving a median read length of 25 bp, compared to 44 bp for typical bone-derived shotgun aDNA in a previous African aDNA study using the same laboratory pipeline4. The second is that unusually high damage rates were observed in the interior of the DNA molecules from hair, while unusually low damage rates were seen in the exterior of the molecules. For instance, at the interior 10th bp from the 5’ end, damage rates were 10% compared to 1% from typical bone-derived aDNA. While at the exterior, the 1st bp from the 5’ end showed damage rates of 15%, compared to on average of 27%22 from typical bone-derived aDNA. These patterns are consistent with high degradation of DNA fragments in hair through intense sun exposure, already during the lifetime of the individual, which may result in hair containing largely denatured single-stranded DNA fragments, as opposed to the more typically intact double-stranded fragments preserved in bone samples.
We find the ratio of mitochondrial to nuclear DNA in the hair-derived DNA library to be relatively high (with a ratio at 224 at length cut-off 25 bp, Table S1), compared to typical rates with other tissues; for example the ratio is at 110 on average for petrous bones in a previous study using the same laboratory pipeline4. We investigated whether there is a notable difference in terms of aDNA preservation in nuclear DNA and mitochondria from the hair material. Specifically, we examined if the two idiosyncratic features of hair aDNA we describe above apply to both nuclear and mitochondrial DNA from hair. In Fig. 2 and Fig S2, we compared the read length distribution and average base substitution rates of reads mapped to the complete genome (i.e., nuclear and mitochondrial), the nuclear genome, and the mitochondrial genome. We find that both nuclear and mitochondrial DNA have high base substitution rates in the interior of sequence reads (Fig S2a, c), but reads mapped to mitochondria are relatively longer than reads mapped to autosomes (Fig S2b, d).
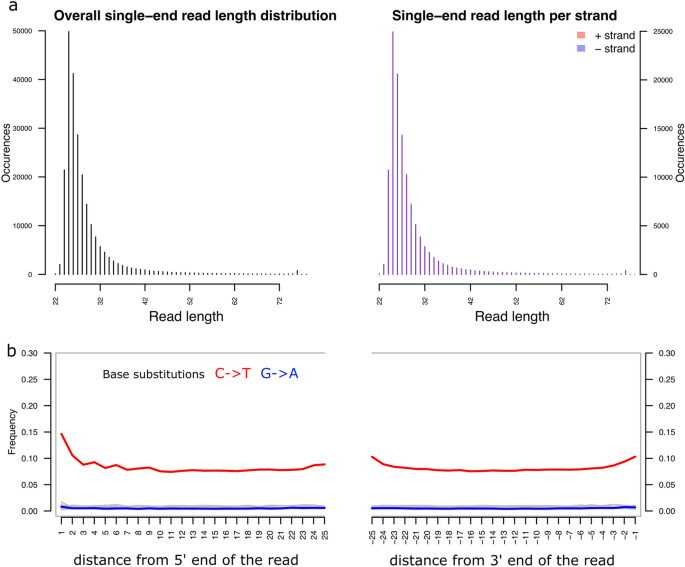
Characteristics of aDNA fragments from the hair sample. (a) Length distribution of shotgun sequencing reads mapped to the whole genome using read length filter cut-off at 25 bp in the step of adaptor removal. (b) High C-to-T substitution rates in the interior of aDNA fragments.
Given the tenfold reduction of mapped reads after applying a mapping quality filter (Table 1), we examined if the two features of hair aDNA observed here resulted from the mapping quality filter. We find that our mapping quality filter did not have a notable effect on the two features of ultra-short DNA fragment enrichment and high interior aDNA damage pattern (Fig S3).
Given the success of SNP capture techniques for poorly preserved human DNA23, we also performed SNP capture for our hair-derived aDNA library. However, SNP capture did not provide an improvement over shotgun sequencing. Instead, we found the base substitution rates in the exterior and interior of reads are substantially lower in capture data, in comparison to the rate distribution in shotgun data (Fig. S1, Table S1), corroborating the fact of high contamination rate in the capture data (42 ± 3% as estimated by AuthentiCT), likely due to capture preferentially targeting molecules without damage (due to more effective hybridization) than with damage. In addition, longer molecules are preferentially captured over short molecules.
Genetic affinity to early eastern African pastoralists
We performed Principal Components Analysis (PCA) and Outgroup-f3 (Figs. 2, 3) to investigate the genetic ancestry of the individual (Sudan_Kadruka1_4000BP) from whom our hair sample derived, utilizing 3336 mapped reads overlapping with SNP positions from the Shotgun data, after read length filter at 25 bp of Sudan_Kadruka1_4000BP (Table S2). To maximise the resolution given the extremely low coverage and low number of called alleles, we used high-coverage modern African genomic data from the SGDP24 and the HGDP25, which includes all SNPs in 1240 k panel, instead of the commonly used Human Origin array data26, for calculating Principal Components (PCs). We projected ancient Africans and ancient Near Easterners on the background of modern African groups25. Although the number of available populations in SGDP and HGDP is limited, we observe clear separations of African populations from different regions, with eastern/northern, southern and western African populations falling into the right, left and top corner of PC1/PC2 space, respectively.
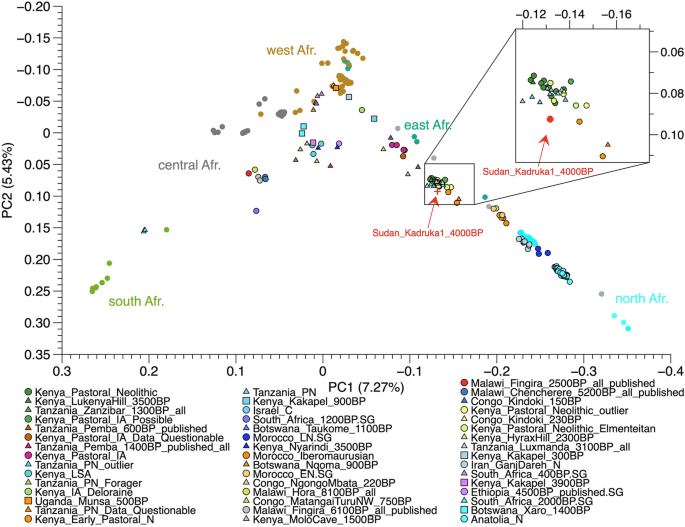
Genetic ancestry of Sudan_Kadruka1_4000BP. Principal Component Analysis (PCA) of African populations. We project Sudan_Kadruka1_4000BP into an African PCA (Table S3) with PCs calculated from modern Africans in SGDP24. We use block jackknife strategy (taking-one-chromosome-out)26 for error bar calculation of Sudan_Kadruka1_4000BP ‘s location on the PCA.
The PCA shows Sudan_Kadruka1_4000BP located close to previously published early pastoralists in eastern Africa4,5, such as Kenya_EarlyPastoral_N (3800–4000 calBP) and Kenya_Pastoral_Neolithic (1500–3000 calBP). Kenya_EarlyPastoral_N is a group of two pastoralist individuals dated to the early stage of the eastern African Pastoral Neolithic, both of whom are genetically derived from admixture between two early northeastern African-related ancestries from Sudan and Northern Africa/Levant5. To estimate the level of noise resulting from the sparsity of our SNP data, we computed a standard error for the projected PCs of Sudan_Kadruka1_4000BP using a block jackknife approach. Specifically, we computed pseudo-values by deleting each chromosome of the genotype data in turn and then used the resulting estimates from the remaining data as input for the weighted jackknife calculation27. We find the standard errors of Sudan_Kadruka1_4000BP to be relatively small compared to overall genetic variation within Africa, which gives us confidence that the location calculated from the full data (Fig. 2a) is robust.
Despite the sparsity of the data, the PCA analyses conducted here clearly suggest a very close genetic relationship between Sudan_Kadruka1_4000BP and ancient eastern African pastoralist populations. To corroborate this finding, we also computed allele sharing rates with ancient populations from the Levant and Africa and present-day African populations at genomic sites where Sudan_Kadruka1_4000BP differs from the chimpanzee reference genome via outgroup-f3 (Sudan_Kadruka1_4000BP, population X; Chimpanzee). Figure 4a shows that Sudan_Kadruka1_4000BP shares the highest genetic affinity with ancient Levantine groups, ancient northern and Eastern Africans and modern Africans from northern Sahara and the Horn of Africa. We computed pairwise comparisons employing f4 (Sudan_Kadruka1_4000BP, Kenya_EarlyPastoral_N; population X, Chimp) to validate the close PC location between Sudan_Kadruka1_4000BP and Kenya_EarlyNeolithic_N. Consistent with PCA location, f4-statistic result confirms the genetic cladality between Sudan_Kadruka1_4000BP and Kenya_EarlyPastoral_N given that none of the tested populations breaks cladality significantly, suggesting that these two individuals are indistinguishable in terms of allele frequencies (Fig. 4b). Additionally, we show that in f4 (Kenya_EarlyPastoral_N, population X; Sudan_Kadruka1_4000BP, Chimp) results (Fig. 4c) that all tested ancient and modern African populations are either significantly positive (suggesting that they are less close to Kenya_EarlyPastoral_N compared to Sudan_Kadruka1_4000BP) or overlapping with zero, indicating equal genetic distance to both.

Genetic affinity with ancient African pastoralists. (a) Outgroup f3(Sudan_Kadruka1_4000BP, population X; Chimp). (b) f4(Sudan_Kadruka1_4000BP, Kenya_EarlyPastoral_N; population X, Chimp). (c) f4(Kenya_EarlyPastoral_N, population X; Sudan_Kadruka1_4000BP, Chimp). Population X includes published ancient African and Near Eastern populations and modern African populations from SGDP and HGDP data sets (Table S3). We plot two standard error bars for f3 and f4 statistics shown here and highlight statistically significant tests (Z-score > 3) in red color.